Authour/Curator: Aviral Vatsa PhD MBBS
This post is in continuation with posts on basics of NO metabolism and its effect on physiology. Other topics are covered under the following posts.
Nitric oxide plays wide variety of roles in cardiovascular system and acts as a central point for signal transduction pathway in endothelium. NO modulates vascular tone, fibrinolysis, blood pressure and proliferation of vascular smooth muscles. In cardiovascular system disruption of NO pathways or alterations in NO production can result in preponderance to hypertension, hypercholesterolemia, diabetes mellitus, atherosclerosis and thrombosis. The three enzyme isoforms of NO synthase family are responsible for generating NO in different tissues under various circumstances. The endothelial NOS (eNOS) is expressed in endothelial cells, the inducible NOS (iNOS) is expressed in macrophages and neuronal NOS (nNOS) is expressed in certain neurons and skeletal muscle. Although the basic mechanism of action for NO production is the same for all three NOS isoforms, yet deficiencies of each one of them manifest differently or with varying severity in the body e.g. eNOS deficiency might lead to hypertension, more severe form of vascular injury to cerebral ischaemia and more severe form of atherosclerosis induced by hypercholesterolemic diet whereas nNOS deficiency might show less severe form of vascular injury to cerebral ischaemia and absence of iNOS might lead to reduced hypotension in septic shock.
Reduction in NO production is implicated as one of the initial factors in initiating endothelial dysfunction. This reduction could be due to
reduction in eNOS production
reduction in eNOS enzymatic activity
reduced bioavailability of NO
eNOS production is increased by physiological sheer stress on endothelial cells resulting from normal flow of blood along the arterial walls. Alterations in fluid sheer stress patterns e.g due to arterial constriction has been shown to have detrimental effect on eNOS production in endothelial cells. eNOS production is decreased by LDL, angiotensin II and TNF alpha. eNOS is tightly coupled enzyme and its activity can be significantly reduced by reduction in availability of cofactors and substrates, and by competitive inhibitors such as ADMA. Furthermore, uncoupling of eNOS can result in increased production of reactive species of both oxygen (superoxide) and nitrogen (peroxinitrite), which inturn can further reduce eNOS bioavailability. A range of therapeutic targets aim at increasing bioavailability of eNOS and they are summarised here.
Increased production of ROS and peroxinitrite is associated with endothelial dysfunction. Coronary heart disease risk factors may increase NOS mediated ROS formation and peroxinitrite formation. Such risk factors are associated with decreased NO production levels in the vasculature. However, recent data suggests that reduction in bioavailable NO levels in the arteries could be due to increased local oxidative stress rather than reduction in basal NO production.
Oxidation dependent mechanisms have been implicated in endothelial dysfunction. Oxidized low density lipo-proteins (oxLDL) play an important role in early endothelial dysfunction and hence early atherosclerosis (see figure below)
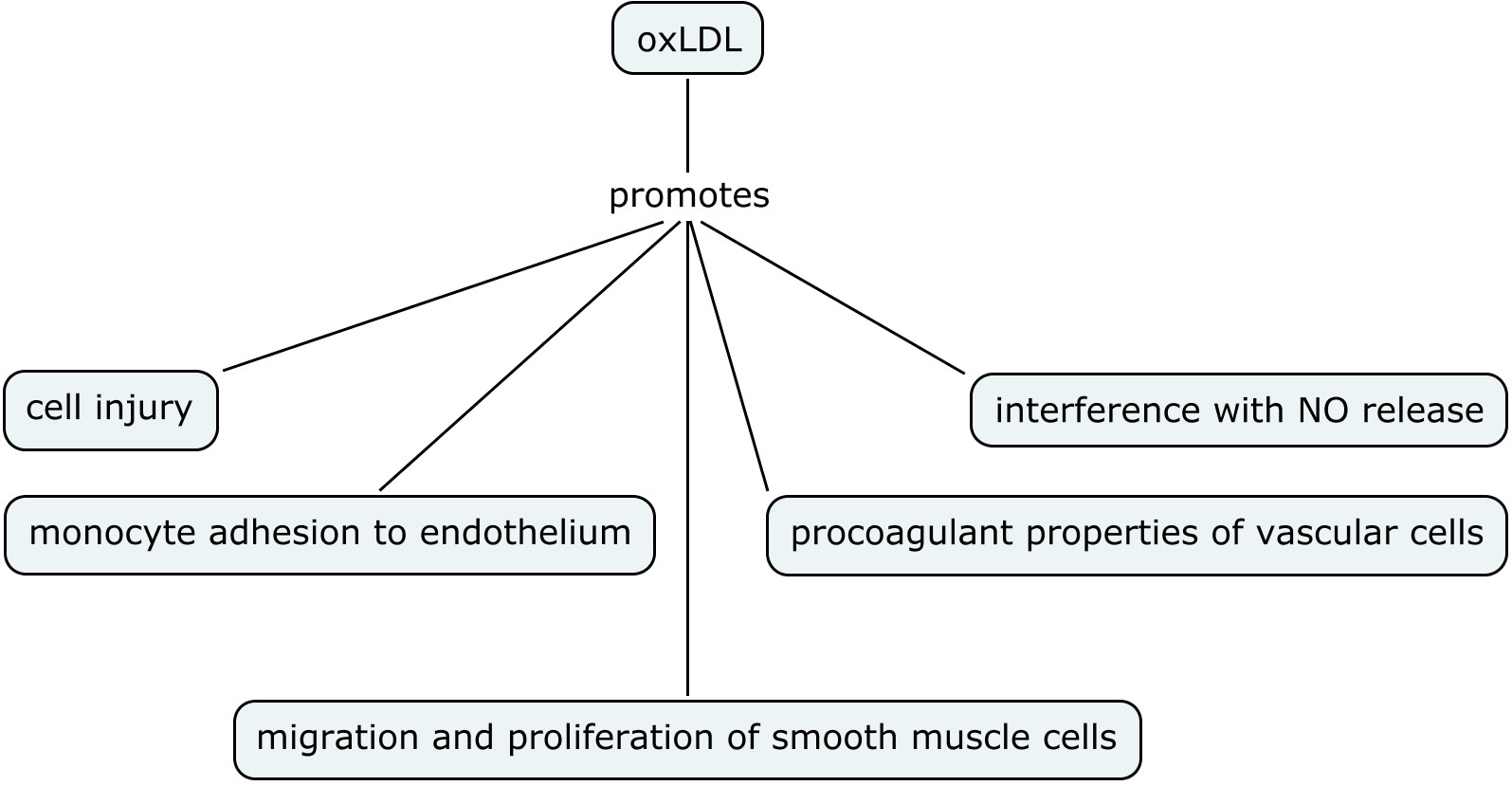
oxLDL can uncouple eNOS and reduced uptake of L-ariginine that can lead to production of superoxide radical oxygen. OxLDL can interfere with NO production and lead to altered NO signalling in the vascular endothelium. In addition, different arteries can be affected differently by these physiological changes e.g. oxLDL affects carotid artery and not the basilar artery thereby implying that intracranial arteries might be protected from endothelium-mediated oxidative injury and hence atherosclerosis. And finally NO can modulate oxidation mediated apoptotic signals in the vessel wall. Hence atherosclerosis can result from the derangement of fine imbalance between NO bioavailability and local oxidative stress.
Therapeutic targets:
There are various pathways being targeted to modulate the bioavailability of eNOS and NO such as
Recoupling of eNOS to cofactors and substrates
Modulation of eNOS activity by genomic and non genomic mechanisms e.g. by statins, ACE inhibitors, angiotensin II receptor blockers, calcium channel blockers , KLF2 modulators
The suppression of inflammatory signalling pathways by PPAR-α activation
Modulation of caeviolin mediated endocytosis and thus dissociation of eNOS from caevolin
The above list is not exhaustive, but this post here summarises recent developments in therapeutic targets in NO /eNOS regulation.
Based on:
Nitric Oxide and Pathogenic Mechanisms Involved in the Development of Vascular Diseases
Claudio Napoli and Louis J. Ignarro
Arch Pharm Res Vol 32, No 8, 1103-1108, 2009 , DOI 10.1007/s12272-009-1801-1

No comments:
Post a Comment